



Next: Contents
Contents
Atomistic Modeling of Biomolecular Interactions
New Mexico Adventures in
Supercomputing Challenge
Final Report
April 7, 2004
026
Eldorado High School
Team Members
Tom Dimiduk
Jeff Dimiduk
Daniel Appel
Ryan Shea
Teacher
David Dixon
Project Mentor
Susan R. Atlas
Executive Summary
Molecular motors are a group of biological proteins, which are critical to life because they are responsible for all transport within cells. They are the primary mechanism for converting chemical energy to mechanical work at very small scales. There are numerous subfamilies of different proteins, but they all basically use ATP (adenosine triphosphate) molecules as an energy source to move 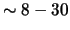 nanometers along a microtubule. Understanding how these proteins work can lead to new therapies for cancer and muscular disorders. A better understanding of how these proteins work could also make it possible to incorporate these proteins into nanoscale biomimetic devices.
The size of molecular motor proteins makes it impossible to directly observe their function, so scientists recently began to attempt to use atomistic simulations to model the protein structure and function. For atomistic modeling, each atom is represented as a particle in space with attributes such as position, velocity, and charge. The modeling is done by approximating forces between particles, and changing the position and velocity of each particle as a result of these forces, (
nanometers along a microtubule. Understanding how these proteins work can lead to new therapies for cancer and muscular disorders. A better understanding of how these proteins work could also make it possible to incorporate these proteins into nanoscale biomimetic devices.
The size of molecular motor proteins makes it impossible to directly observe their function, so scientists recently began to attempt to use atomistic simulations to model the protein structure and function. For atomistic modeling, each atom is represented as a particle in space with attributes such as position, velocity, and charge. The modeling is done by approximating forces between particles, and changing the position and velocity of each particle as a result of these forces, (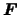 = m
= m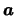 ), for each unit of time. These calculations become very computationally challenging when modeling biophysical systems because proteins exist in aqueous solutions containing many thousands of atoms and ions that need to be individually modeled in addition to modeling the protein itself.
The particular part of the molecular motor dynamics simulation that we worked on was the calculation of long-range electrostatic interactions. In the specific case of long-range forces, the behavior of charged particle interactions requires a sum over a prohibitively large number of particles. We use a periodically repeated unit cell to simplify calculations without introducing artificial boundary effects. We further simplify the calculations by using Ewald summation, a technique in which a conditionally convergent sum is simplified to a real space sum, and a reciprocal (or Fourier) space sum. We then are able to increase the efficiency of our calculations by using Particle Mesh Ewald, a method that allows us to use Fast-Fourier Transforms to calculating the Fourier space term.
Computational efficiency is of utmost importance because modeling molecular motor systems containing many thousands of atoms is computationally intensive. Our Fortran90 code successfully combined modules to calculate the complementary error function, Cardinal B-Splines, and mesh interpolation in order to implement the Particle Mesh Ewald method. With this, we calculated the real and Fourier of the Particle Mesh Ewald method, and could calculate long-range interactions for the many thousands of particles needed in molecular motor biophysical simulations.
), for each unit of time. These calculations become very computationally challenging when modeling biophysical systems because proteins exist in aqueous solutions containing many thousands of atoms and ions that need to be individually modeled in addition to modeling the protein itself.
The particular part of the molecular motor dynamics simulation that we worked on was the calculation of long-range electrostatic interactions. In the specific case of long-range forces, the behavior of charged particle interactions requires a sum over a prohibitively large number of particles. We use a periodically repeated unit cell to simplify calculations without introducing artificial boundary effects. We further simplify the calculations by using Ewald summation, a technique in which a conditionally convergent sum is simplified to a real space sum, and a reciprocal (or Fourier) space sum. We then are able to increase the efficiency of our calculations by using Particle Mesh Ewald, a method that allows us to use Fast-Fourier Transforms to calculating the Fourier space term.
Computational efficiency is of utmost importance because modeling molecular motor systems containing many thousands of atoms is computationally intensive. Our Fortran90 code successfully combined modules to calculate the complementary error function, Cardinal B-Splines, and mesh interpolation in order to implement the Particle Mesh Ewald method. With this, we calculated the real and Fourier of the Particle Mesh Ewald method, and could calculate long-range interactions for the many thousands of particles needed in molecular motor biophysical simulations.
Next: Contents
Contents
Thomas G Dimiduk
2004-04-15